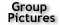Tracking single molecules in living cells provides a direct way to probe the kinetics of their interactions with other cellular components, and is particularly useful to characterize unsynchronized dynamic events. Despite previous successes in single-molecule imaging in live bacterial cells, imaging single fluorescent proteins remains challenging in the nuclei of living mammalian cells, due to their larger size and higher fluorescence background. We developed a novel fluorescence microscopy technique that combines selective plane illumination with a vertical arrangement of illumination and detection objectives. In this new geometry (Fig. 1), a disposable mirror reflects the light sheet into a horizontal plane close to the sample surface, thus allowing horizontal sectioning of the cells and achieving single fluorescent protein imaging in live mammalian cells. We name this technique reflected light sheet microscopy (RLSM).
Our technique possesses the advantage of superior signal-to-background ratio, fast image acquisition speed and millisecond time resolution, low photobleaching rate and reduced phototoxicity, as well as easy optical sectioning capability. The reflected light sheet configuration also allows individual, normal-sized adherent cells to be imaged, as compared to other selective plane illumination methods that could only be applied to embryos or cellular spheroids much larger in size. Lastly, the single-molecule tracking capability of RLSM allows us to directly monitor in vivo protein dynamics without the need for additional calibrations or corrections that are commonly associated with other techniques such as FRAP and FCS.
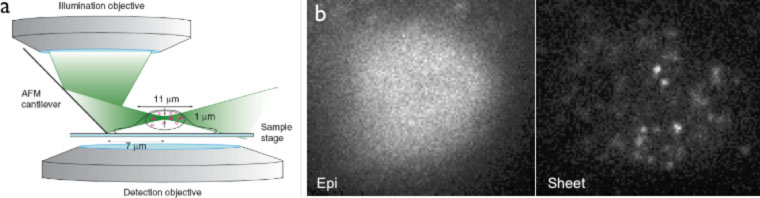
Fig. 1. Visualization of single fluorescent protein molecules in a mammalian nucleus by RLSM. (a) Scheme of the reflected light-sheet principle (not drawn to scale). A laser beam is focused by an objective to form a vertical light sheet that is reflected by 90° off an AFM cantilever next to a cell in a petri dish. Fluorescence is detected by a second high-NA objective. Three-dimensional optical sectioning is achieved through vertical displacement of the sample. (b) A MCF-7 cell expressing mEos2-NFY-A is activated with 405 nm and imaged with 560 nm under epi illumination (left) and RLSM (right), demonstrating the drastic improvement in SBR achievable by RLSM.
We demonstrate the potential of our new microscopy method by directly monitoring the binding properties of fluorescently labeled glucocorticoid receptors (GR) to DNA (Fig. 2a and b). GR is a transcription factor that localizes mostly to the cytoplasm in the absence of hormone, but forms homodimers and translocates into the nucleus upon binding to hormone. Previous studies have shown that dimeric GR binds directly to DNA at regulatory sequences, while the monomer can be indirectly recruited to DNA by other DNA-bound protein complexes. The mode of DNA interaction defines whether the target gene is activated or repressed. Using RLSM, we measured the DNA-bound fraction of GR and determined the residence times on DNA of the various oligomerization states of GR, thereby permitting us to resolve the transcription factor's different modes of DNA binding. We also demonstrated the capability of RLSM for two-color single-molecule imaging (Fig. 2c) by directly observing the spatio-temporal co-localization of GR and its coactivator GRIP1. The imaging technique we have developed could be generally applied to study the dynamics of a variety of key biomolecular players inside the living mammalian nucleus.
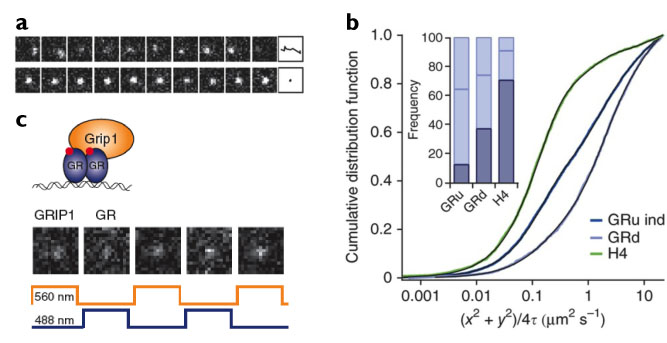
Fig. 2. Characterizing mammalian transcription factor dynamics in vivo with RLSM. (a) Single-molecule tracking of a fast-diffusing mEos2-GR molecule (top) and of a DNA-bound molecule (bottom) in presence of 100 nM dexamethasone at 10-ms time resolution. Far right, identified traces. (b) Cumulative distribution of squared displacements of mEos2-histone H4 (H4) and mEos2-GR with (GRd) or without (GRu) 100 nM dexamethasone induction. Inset, fractions of molecules exhibiting slow effective diffusion (corresponding to DNA-bound fraction, dark blue) and of molecules exhibiting two fast effective diffusion components (light blue). (c) Two-color imaging of GR-GRIP1 colocalization. TagRFP-T-GR (blue trace) and EGFP-GRIP1 (orange trace) were alternately excited with 50-ms exposure time.
In addition, we combined RLS illumination with super-resolution microscopy (SRM), and applied RLS-SRM to image the spatial organization of RNAP II molecules inside the mammalian nucleus, which has long been proposed to heterogeneously occur in discrete foci termed ‘transcription factories’ (Fig. 3A). Leveraging on the blinking photophysics of rhodamine-based dyes such as TMR, we also developed a density-based clustering algorithm that pools multiple localizations in super-resolution images based on their spatial and temporal proximity, so as to be able to accurately count the copy number of RNAP II molecules in transcription foci (Fig. 3B).
Fig. 3. 'Transcription factories' hypothesis and strategy for molecular counting. (A) Models of spatial distribution of RNAP II in mammalian nucleus, in which transcription is carried out by either individual RNAP II molecules (top) or multiple molecules clustered into spatially discrete 'factories' (bottom) that pull together genes to be transcribed. (B) Flow chart of spatio-temporal clustering analysis based on spatial and temporal nearest neighbor distance (NND) thresholding of TMR blinking events, thus enabling accurate counting of RNAP II molecules in transcription foci.
Using RLS-SRM, we quantified the global extent of RNAP II clustering inside the mammalian nucleus. We found that the majority (< 70%) of the transcription foci originate from single RNAP II molecules, and no significant clustering between RNAP II molecules is detected within the length scale of the reported diameter of 'transcription factories' (Fig. 4). Colocalization measurements of RNAP II molecules equally labeled by two spectrally distinct dyes confirmed the primarily unclustered distribution, arguing against a prevalent existence of 'transcription factories' in the mammalian nucleus as previously proposed. Our study not only provides clear and convincing answers to this long-standing controversy, but also paves way for quantitative mapping and stoichiometric characterization of other biomolecular species deep inside mammalian cells.
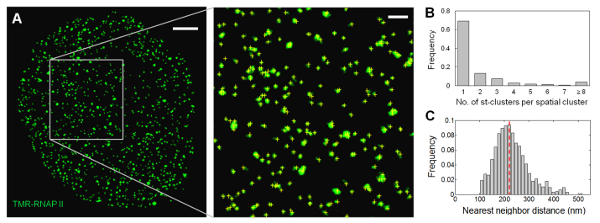
Fig. 4. Spatial organization of RNAP II molecules shows no significant clustering. (A) Distribution of SNAP-RNAP II molecules in a thin optical section of the nucleus of a fixed U2OS cell labeled with TMR. Inset shows a zoomed-in area, where single transcription foci are discernible; yellow crosses indicate the centroid position of the st-clusters identified. Scale bar: 2 μm; inset: 500 nm. (B) Distribution of the number of st-clusters in transcription foci indicates that at least 70% of the foci consist of only one RNAP II molecule (n = 4465). (C) Distribution of spatial NND for transcription foci shows that the majority of the RNAP II molecules do not associate with each other within the reported diameter of 'transcription factories' (40-130 nm). Dotted line indicates mean.
References:
- Gebhardt, J. Christof M.; Suter, David M.; Roy, Rahul; Zhao, Ziqing W.; Chapman, Alec R.; Basu, Srinjan; Maniatis, Tom; Xie, X. Sunney. "Single-Molecule Imaging of Transcription Factor Binding to DNA in Live Mammalian Cells," Nat Methods 10, 421-426 DOI:10.1038/NMETH.2411 (2013)
- Zhao, Ziqing W.; Roy, Rahul; Gebhardt, J. Christof M.; Suter, David M.; Chapman, Alec R.; Xie, X. Sunney. "Spatial organization of RNA polymerase II inside a mammalian cell nucleus revealed by reflected light-sheet superresolution microscopy," PNAS 111, 681-686 DOI:10.1073/pnas.1318496111 (2014)
|