The conformational dynamics of a biomolecule is crucial to its biological functions. Single molecule techniques are particularly informative about conformational dynamics because these motions are usually unsynchronized among the molecules. There are two types of conformational changes, one is the spontaneous, or thermal activated fluctuation, the other is a nonequilibrium process driven by substrate binding or chemical reactions.
Fluorescence Resonance Energy Transfer (FRET) technique is the most widely used fluorescent method for studying conformational changes. Single molecule FRET was first demonstrated by Ha and Weiss [1]. However, it is only sensitive to the distance change on the order of nanometers. We have developed two methods based on single-molecule electron transfer [2] and motion hindering in the excited stated [3], which are complementary to FRET.
Single-Molecule Conformational Dynamics Probed by Electron Transfer
We studied a flavin-enzyme, a flavinreductase (Fre), which catalyzes the reduction of a flavin by NADH (Fig. 1A). In contrast to the free flavin fluorescence with a mono-exponential decay with a long lifetime, the decay of Fre-bound flavins is much faster and is multi-exponential, which have been attributed to quenching of the excited state by ET from nearby residues tyrosines. There are three tyrosines around FAD. Through site-directed mutagenesis, we proved that tyrosine35, the nearest to flavin, is the major quencher. We use excited state electron transfer (ET) to probe the subtle structure change within the intact protein of Fre.
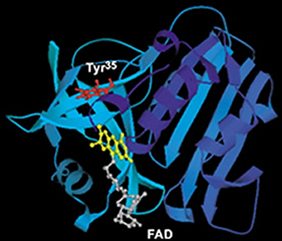 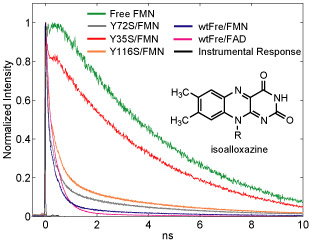
Fig. 1. Fre structure (left) and fluorescence decays (right) of the wide type and mutants of Fre. We replaced the three tyrosines (Y) individually with a serine (S) and generated Y35S, Y72S, and Y116S— by site-directed mutagenesis. Drastic increase of fluorescence lifetime was only observed for the Y35S complex (red curve), proving the quenching by tyrosine Y35 [2].
Another system is a protein complex formed between fluorescein (FL) and monoclonal
antifluorescein 4-4-20 (anti-FL). This complex is highly stable, allowing long-time observations at
the single-molecule level. Fig.2 shows its crystal structure [6]. Fluorescein is the ET acceptor and a nearby tyrosine donor is the donor. The shortened fluorescein fluorescence lifetime by the tyrosin is shown Fig. 2.
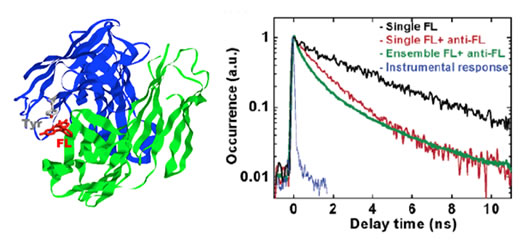
Fig. 2. (left) Fluorescein (FL) and anti-FL complex with the electron transfer donor and acceptor, Tyr37 and FL, respectively. (right) Monoexponential fluorescence lifetime decay for a single FL molecule (black curve), multiexponential fluorescence decay for the FL/anti-FL complex at both ensemble (green curve) and single- molecule (red curve) levels, and the instrumental response function (blue curve) [5].
The energy diagram of the excited state Et is shown in Fig. 3. The fluorescence lifetime t is determined by the radiative rate γ relaxation rate, the fluorescence lifetime t = 1/(γ + kEt). The Et rate is highly dependent on the edge to edge distance between the electron donor and acceptor following kEt= k0*exp(- β R). Therefore, the fluctuation of the donor-acceptor distance R results in the fluctuation of the fluorescence lifetime t. The scaling factor b is around 1.0~1.4 Å-1 for proteins, which makes Et a distance dependent probe for the subtle motions on angstrom scale. The amino acid residues tyrosine and tryptophan or DNA base Guanine can serve as natural Et donors. A typical Et acceptors can be natural chromophores such as flavin, or artificial dyes such as rhodamines.
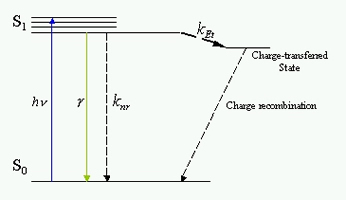 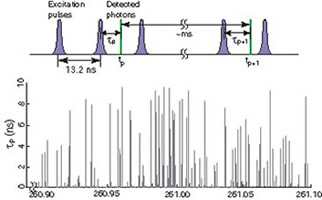
Fig. 3. Probing single molecule conformational dynamics by electron transfer, on a photon-by-photon basis [2].
We use a confocal microscope to detect the fluorescence signal from a single molecule. Different from the conventional time correlated single photon counting, for each detected fluorescence photon with index, p, we record both the delay time respective to its excitation pulse, Τp, and the chronological arrival time tp (Fig. 3). Instead of binning the detected photon to calculate the fluorescence lifetime,
the lifetime correlation function C(t) can be calculated with the novel photon-by-photon
correlation method. Such a method provides dynamic information a broad range of time scale, particularly the long time scales not accessible by molecular dynamics simulations.
Equilibrium Conformational Dynamics Explained by the Generalized Langevin Equation
We found in the two systems described above that the distance between an electron transfer donor
and an acceptor was found to take place at a broad range of time scales, ranging from 10ms to 10s
(Fig. 4) [2,5]. This equilibrium conformational fluctuation is a general phenomenon that occurs in
Other systems, even in MD simulations (5). Such conformational fluctuations at a broad range of time
scales result from the flexible but glassy nature of enzymes as biopolymers, and result in fluctuations in the rates of enzymatic reactions. The physical picture to explain the observation is that a protein
molecule is a “sluggish” system in which one degree of freedom (say, x) is strongly coupled to other
degrees of freedom. A change in x requires collective motions of the other degrees of freedom.
This results in a long time memory effect, a characteristic of non-Markovian behavior. We have provided a mathematical reduction of this picture by the generalized Langevin equation [6].
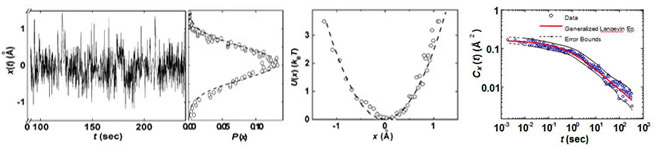
Fig. 4 (A) Segment of the donor–acceptor distance, x(t) trajectory with the corresponding probability density function P(x) with a Gaussian fit. (B) Harmonic potential of mean force, U(x) = −kBTln[P(x)]. (C) Autocorrelation of the donor–acceptor distance in (A) that shows the broad range of time scale distance fluctuation, which can be fit well with the generalized Langevin equation [5].
Probing Non-equilibrium Conformational Motions by Fluorescence Intensity Changes of a Hindered Fluorophore
Functionally important conformational motions of macromolecules, such as enzymes, are often
nonequilibrium processes driven by substrate binding or chemical reactions. Unlike the equilibrium
fluctuations that are ubiquitous for any two points inside a macromolecule, functionally important
conformational changes are often designed and evolved by nature.
For example, DNA polymerase, is a molecular copy machine for DNA, whose structure resembles a palm
and fingers holding a primed DNA. According to the crystal structure, upon binding of the substrate
dNTP, it undergoes a conformational change, known as closing of the fingers’ domain, as shown in
the animation extrapolated from the structures with and without the substrate (Fig. 5).
We designed an assay to probe this motion in real-time based on fluorescence intensity changes of a
fluorophore (Cy3) within the protein (DNA polymerase). When the protein conformation hinders
torsional motions of the dye, the fluorescene intensity is increased. Similar increases of fluorescence
intensity was seen in solutions when the viscosity of the solvent is increased (Fig. 5).
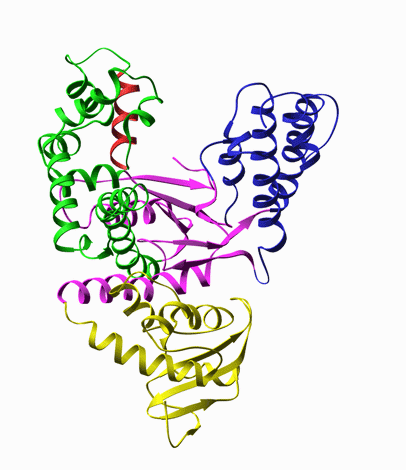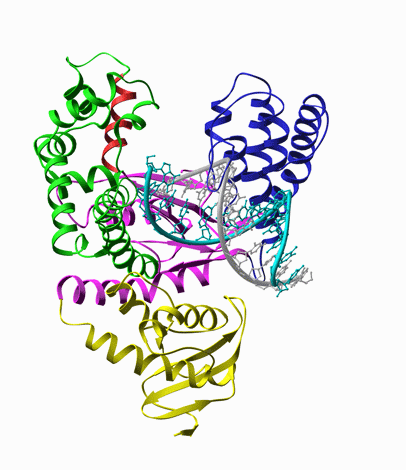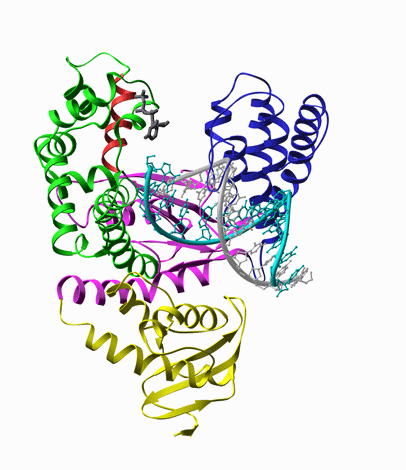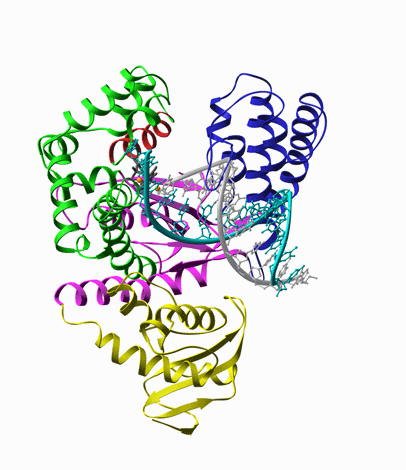 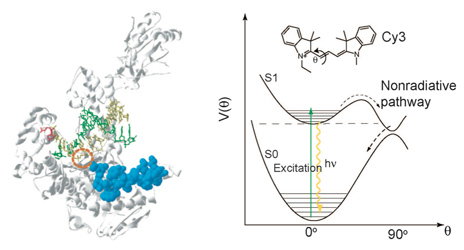
Fig. 5. Left: Closing of DNA Polymerase's fingers' domain, according to the x-ray structures with and without the dNTP substrate [8]. Middle: T7 RNA polymerase bound on a primed DNA duplex that is labeled with a Cy3 dye molecule (red circle). Right: Fluorescence intensity of Cy3 is increased when the nonradiative decay rate of Cy3 is decreased due to hindered torsional motions of the excited state of Cy3 molecule.
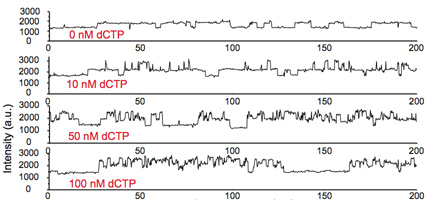
Fig. 6. Fluorescence intensity trajectories of a surface-immobilized primed DNA duplex that is labeled with a Cy3 dye in the presence of DNA polymerase in solution [3].
Figure 6 shows, at zero substrate concentration (first time trace), the intensity switches between two levels: the lower level corresponds to free DNA and the higher level corresponds to the polymerase bound on DNA, hindering the torsional motion of Cy3. When a matched substrate dCTP is introduced, the fluorescence intensity increased to yet another higher level, due to the conformational changes associated with the closing of the fingers’ domain. The subsequent polymerization reaction is blocked such that the dCTP dissociates and the same process can be monitored repeatedly. As a control experiment, when the exo-nuclease part of the polymerase (blue part) is removed, freeing the hindered Cy3 motion, the intensity increases no longer occur.
References
[1] S. Weiss, Nature Structural Biology, 2000, 7, 724-729.
[2] Yang, H.; Luo, G.; Karnchanaphanurach, P.; Louie, T.-M.; Rech,I.; Cova, S.; Xun, L.; Xie, X. S. Protein Conformational Dynamics Probed by Single-Molecule Electron Transfer. Science 2003, 302, 262-266.
[3] Luo, Guobin; Wang, Mina; Konigsberg, William H.; Xie, X. Sunney "Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase,"PNAS, 104, 12610-12615 (2007).
[4] M. Whitlow et al., Protein Eng. 1995, 8, 749 (1995),
[5] Min,W.;Luo,G.;Cherayil,B.J.;Kou,S.C.;Xie,X.S.Observation of a Power Law Memory Kernel for Fluctuations within a Single Protein Molecule. Phys. Rev. Lett. 2005, 94, 198302(1)-198302-(4).
[6] Kou, S. C.; Xie, X. S. Generalized Langevin Equation with Fractional Gaussian Noise: Subdiffusion within a Single Protein Molecule. Phys. Rev. Lett.2004, 93, 180603(1)-180603(4).
[7] Luo, Guobin; Andricioaei, Ioan; Xie, X. Sunney; Karplus, Martin "Dynamic Distance Disorder in Proteins Is Caused by Trapping," J. Phys. Chem. B,2006,110, 9363-9367.
[8] Li, Ying; Korolev, Sergey; Waksman, Gabriel. "Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation," EMBO J., 1998, 17, 7514.
|