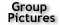Single-cell genomics is important for biology and medicine. However, previous whole-genome amplification (WGA) methods have limited capability to detect copy-number variations (CNVs) and single-nucleotide variations (SNVs). This can be attributed to the low amplification evenness and fidelity by exponential amplification (Figure 1).
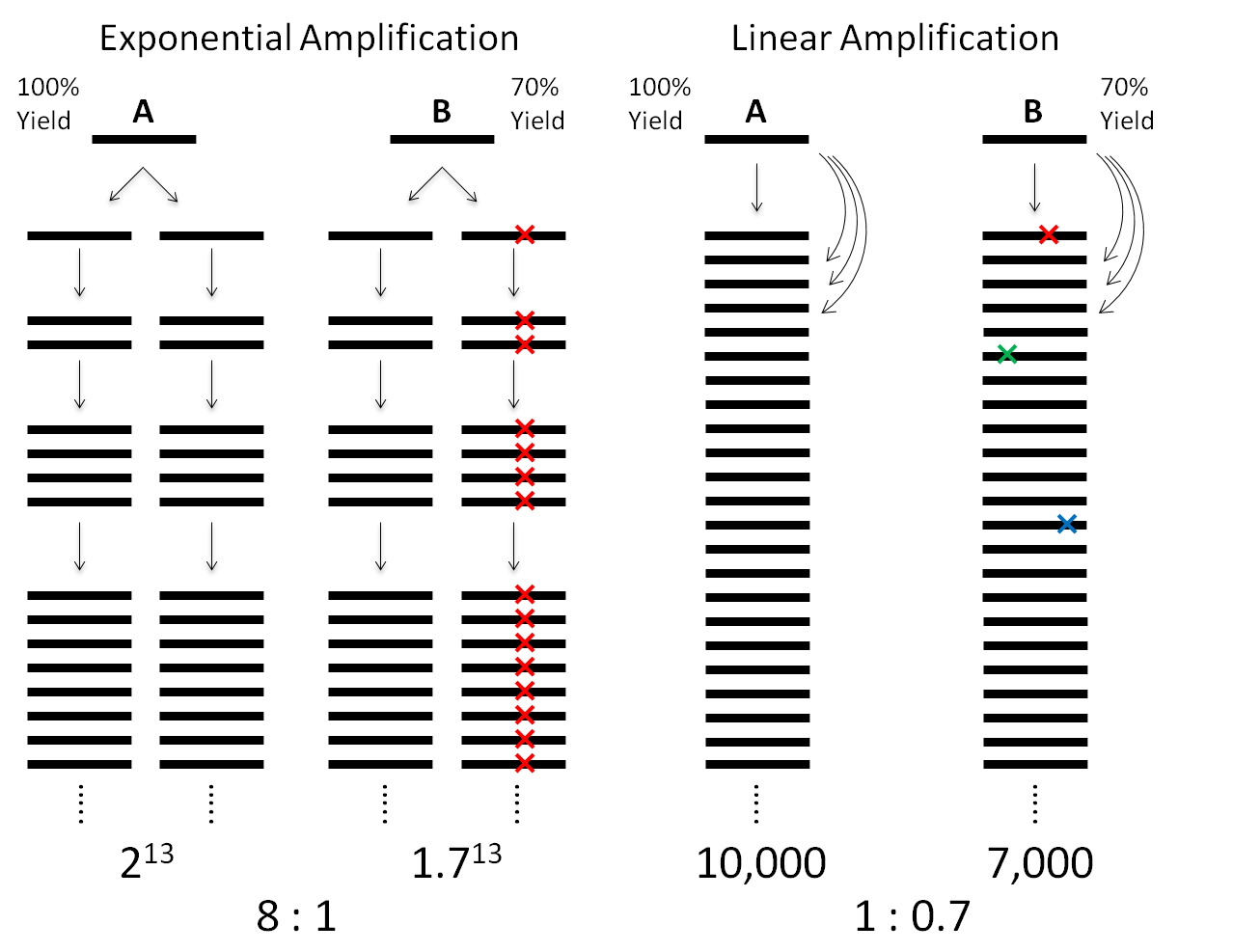
Fig. 1.
Comparison between exponential amplification and linear amplification, assuming the DNA fragments A and B have replication yields of 100 and 70% per round, respectively. For a final amplification factor of ~10,000 of fragment A, exponential amplification results in a ratio of 8:1, hampering the accuracy of CNV detection. In contrast, linear amplification exhibits a ratio of 1:0.7,which is much closer to unity. Linear amplification is also superior to exponential amplification in fidelity. In exponential amplification, a polymerase of the highest fidelity (10−7) replicating the human genome (3 × 109 bp) in the first cycle would give ~300 errors, which will be propagated permanently in the next replication cycles, leading to false-positive SNVs. In contrast, in linear amplification, the errors would appear randomly at different locations in the amplicons and can be easily filtered out.
To overcome the limitations of exponential amplification, we have developed a new single-cell WGA method, Linear Amplification via Transposon Insertion (LIANTI). LIANTI takes advantage of Tn5 transposition and T7 in vitro transcription to linearly amplify genomic DNA fragments from a single human cell (Figure 2).
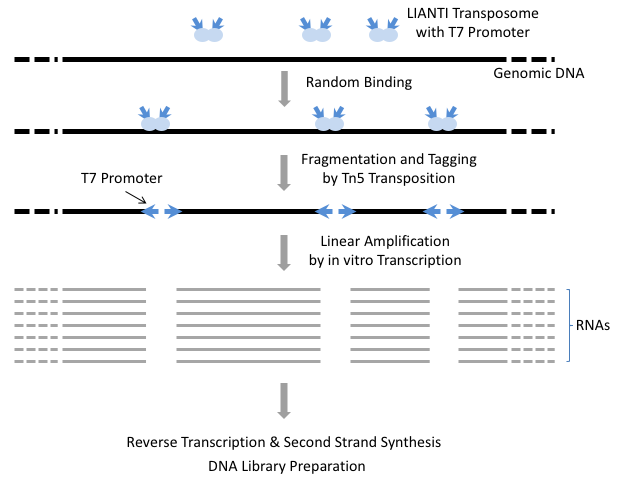
Fig. 2.
The scheme of LIANTI. Genomic DNA from a single cell is randomly fragmented and tagged by LIANTI transposome with T7 promoter, followed by in vitro transcription to linearly amplify each DNA fragment. After reverse transcription and second-strand synthesis, the amplicons are ready for library preparation and sequencing.
LIANTI exhibits the highest amplification uniformity compared to the other methods (Figure 3A, 3B). Together with its unique capability of digital counting to infer fragment numbers based on the mapping coordinates of each amplicon, LIANTI allows accurate detection of single-cell micro-CNVs with kilobase resolution (Figure 3C, 3D).
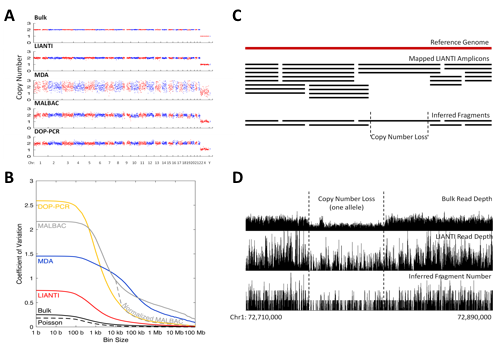
Fig. 3.
LIANTI amplification uniformity and micro-CNV detection. (A) Read depth across the genome with 1-Mb bin size. (B) Coefficient of variation for read depths along the genome as a function of bin size. (C) Principle of digital counting to infer fragment numbers by LIANTI. (D) Example of a 57-kb 2-to-1 copy-number loss detected in a single human cell.
LIANTI also achieves the highest amplification fidelity for accurate single-cell SNV detection (Figure 4A). Furthermore, we show that the predominant cytosine-to-thymine mutations observed in single-cell genomics are often SNV false positives arising from the artifact of cytosine deamination upon cell lysis (Figure 4B).
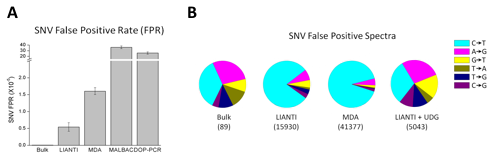
Fig. 4.
LIANTI amplification fidelity and SNV detection. (A) False-positive rates of SNV detection in a single human cell. (B) Spectra of SNV false positives.
Using LIANTI, we directly observed the stochastic firing of DNA replication origins, which differs from cell to cell. We also determined the spectrum and genome-wide distribution of ultraviolet-induced SNVs in a single human cell. In addition, the high precision of micro-CNVs detection and the ability to call individual SNVs in a single cell will allow better genetic screening in reproductive medicine and provide valuable information about how genome variation takes place in cancer and other diseases.
References:
Chen, Chongyi; Xing, Dong; Tan, Longzhi; Li, Heng; Zhou, Guangyu; Huang, Lei; Xie, X. Sunney "Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI)," Science 356, 189-194. DOI:10.1126/science.aak9787 (2017)
|